
B.Pharmacy 7th Semester Notes
UV Visible Spectroscopy
Instrument method of analysis


- E = hν = h c / λ
- Wavelength (λ) = 1 / ū = c / ν
- Wave-number (ū) = 1 / λ = ν / c
- Frequency (ν) = c / λ = c ū
- Velocity (c) = νλ = ν / ū
- 1 Ǻ = 10-1 nm = 10-8 cm
Electromagnetic spectrum

Electronic Excitation by UV/Vis Spectroscopy
 |
| Electronic Excitation by UV/Vis Spectroscopy |
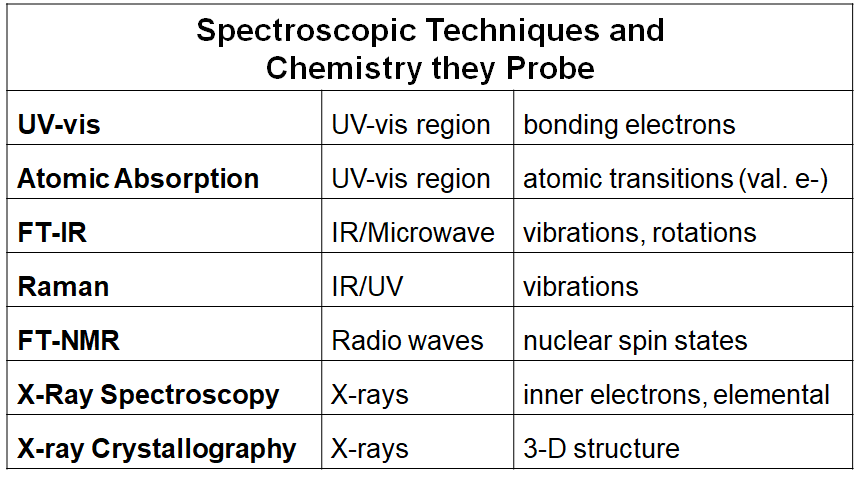 |
Spectroscopic Techniques and Chemistry they Probe |
 |
Spectroscopic Techniques and Common Uses |
Introduction
A. UV radiation and Electronic Excitations
The difference in energy between molecular bonding, non-bonding and anti-bonding orbitals ranges from 125-650 kJ/mole.
This energy corresponds to EM radiation in the ultraviolet (UV) region, 100-350 nm, and visible (VIS) regions 350-700 nm of the spectrum
B. The Spectroscopic Process
- In UV spectroscopy, the sample is irradiated with the broad spectrum of the UV radiation.
- If a particular electronic transition matches the energy of a certain band of UV, it will be absorbed.
- The remaining UV light passes through the sample and is observed.
- From this residual radiation a spectrum is obtained with “gaps” at these discrete energies – this is called an absorption spectrum.

C. Observed electronic transitions
- The lowest energy transition (and most often obs. by UV) is typically that of an electron in the Highest Occupied Molecular Orbital (HOMO) to the Lowest Unoccupied Molecular Orbital (LUMO)
- For any bond (pair of electrons) in a molecule, the molecular orbitals are a mixture of the two contributing atomic orbitals; for every bonding orbital “created” from this mixing (σ, π), there is a corresponding anti-bonding orbital of symmetrically higher energy (σ*, π*)
- The lowest energy occupied orbitals are typically the σ; likewise, the corresponding anti-bonding σ* orbital is of the highest energy
- p-orbitals are of somewhat higher energy, and their complementary anti-bonding orbital somewhat lower in energy than σ*.
- Unshared pairs lie at the energy of the original atomic orbital, most often this energy is higher than π or σ (since no bond is formed, there is no benefit in energy)
- Here is a graphical representation

- From the molecular orbital diagram, there are several possible electronic transitions that can occur, each of a different relative energy:

- Although the UV spectrum extends below 100 nm (high energy), oxygen in the atmosphere is not transparent below 200 nm.
- Special equipment to study vacuum or far UV is required.
- Routine organic UV spectra are typically collected from 200-700 nm.
- This limits the transitions that can be observed:

D. Selection Rules
- Not all transitions that are possible are observed.
- For an electron to transition, certain quantum mechanical constraints apply – these are called “selection rules”.
- For example, an electron cannot change its spin quantum number during a transition – these are “forbidden”.
- Other examples include:
- the number of electrons that can be excited at one time
- symmetry properties of the molecule
- symmetry of the electronic states
- To further complicate matters, “forbidden” transitions are sometimes observed (albeit at low intensity) due to other factors
E. Band Structure
- Unlike IR (or later NMR), where there may be upwards of 5 or more resolvable peaks from which to elucidate structural information, UV tends to give wide, overlapping bands
- It would seem that since the electronic energy levels of a pure sample of molecules would be quantized, fine, discrete bands would be observed – for atomic spectra, this is the case
- In molecules, when a bulk sample of molecules is observed, not all bonds (read – pairs of electrons) are in the same vibrational or rotational energy states
- This effect will impact the wavelength at which a transition is observed – very similar to the effect of H-bonding on the O-H vibrational energy levels in neat samples
- When these energy levels are superimposed, the effect can be readily explained – any transition has the possibility of being observed.

Chromophores
A. Definition
- Remember the electrons present in organic molecules are involved in covalent bonds or lone pairs of electrons on atoms such as O or N
- Since similar functional groups will have electrons capable of discrete classes of transitions, the characteristic energy of these energies is more representative of the functional group than the electrons themselves
- A functional group capable of having characteristic electronic transitions is called a chromophore (color loving)
- Structural or electronic changes in the chromophore can be quantified and used to predict shifts in the observed electronic transitions
B. Organic Chromophores
- Alkanes – only posses σ-bonds and no lone pairs of electrons, so only the high energy σ → σ* transition is observed in the far UV. This transition is destructive to the molecule, causing cleavage of the σ-bond.

- Alcohols, ethers, amines and sulfur compounds – in the cases of simple, aliphatic examples of these compounds the n → σ* is the most often observed transition; like the alkane σ → σ* it is most often at shorter λ than 200 nm. Note how this transition occurs from the HOMO to the LUMO.

- Alkenes and Alkynes – in the case of isolated examples of these compounds the π → π* is observed at 175 and 170 nm, respectively. Even though this transition is of lower energy than σ → σ*, it is still in the far UV – however, the transition energy is sensitive to substitution.

- Carbonyls – unsaturated systems incorporating N or O can undergo n → π* transitions (~285 nm) in addition to π → π*.Despite the fact this transition is forbidden by the selection rules (ε = 15), it is the most often observed and studied transition for carbonyls.This transition is also sensitive to substituents on the carbonyl.Similar to alkenes and alkynes, non-substituted carbonyls undergo the π → π* transition in the vacuum UV (188 nm, ε = 900); sensitive to substitution effects.n → π* transitions (~285 nm); π → π* (188 nm)

C. Substituent Effects
General – from brief study of these general chromophores, only the weak n → π* transition occurs in the routinely observed UV.
The attachment of substituent groups (other than H) can shift the energy of the transition.
Substituents that increase the intensity and often wavelength of an absorption are called auxochromes.
Common auxochromes include alkyl, hydroxyl, alkoxy and amino groups and the halogens.
Substituents may have any of four effects on a chromophore:
- Bathochromic shift (red shift) – a shift to longer λ; lower energy
- Hypsochromic shift (blue shift) – shift to shorter λ; higher energy
- Hyperchromic effect – an increase in intensity
- Hypochromic effect – a decrease in intensity

- Conjugation – most efficient means of bringing about a bathochromic and hyperchromic shift of an unsaturated chromophore:

- Conjugation – AlkenesThe observed shifts from conjugation imply that an increase in conjugation decreases the energy required for electronic excitation.From molecular orbital (MO) theory two atomic p orbitals, ⲫ1 and ⲫ2 from two sp2 hybrid carbons combine to form two MOs ψ1 and ψ2* in ethylene.When we consider butadiene, we are now mixing 4 p orbitals giving 4 MOs of an energetically symmetrical distribution compared to ethylene.

 Extending this effect out to longer conjugated systems the energy gap becomes progressively smaller:
Extending this effect out to longer conjugated systems the energy gap becomes progressively smaller: Similarly, the lone pairs of electrons on N, O, S, X can extend conjugated systems – auxochromes.Here we create 3 MOs – this interaction is not as strong as that of a conjugated π-system.
Similarly, the lone pairs of electrons on N, O, S, X can extend conjugated systems – auxochromes.Here we create 3 MOs – this interaction is not as strong as that of a conjugated π-system. Methyl groups also cause a bathochromic shift, even though they are devoid of π- or n-electrons.This effect is thought to be through what is termed “hyperconjugation” or sigma bond resonance.
Methyl groups also cause a bathochromic shift, even though they are devoid of π- or n-electrons.This effect is thought to be through what is termed “hyperconjugation” or sigma bond resonance.
Structure Determination
A. Dienes
- General FeaturesFor acyclic butadiene, two conformers are possible – s-cis and s-transThe s-cis conformer is at an overall higher potential energy than the s-trans; therefore the HOMO electrons of the conjugated system have less of a jump to the LUMO – lower energy, longer wavelength.Two possible π → π* transitions can occur for butadiene ψ2 → ψ3* and ψ2 → ψ4*The ψ2 → ψ4* transition is not typically observed:
- The energy of this transition places it outside the region typically observed – 175 nm.
- For the more favourable s-trans conformation, this transition is forbidden.
The ψ2 → ψ3* transition is observed as an intense absorption.The ψ2 → ψ3* transition is observed as an intense absorption (ε = 20,000+) based at 217 nm within the observed region of the UV.While this band is insensitive to solvent (as would be expected) it is subject to the bathochromic and hyperchromic effects of alkyl substituents as well as further conjugation.Consider: The effect of substituent groups can be reliably quantified from empirical observation of known conjugated structures and applied to new systems.This quantification is referred to as the Woodward-Fieser Rules for eg.
The effect of substituent groups can be reliably quantified from empirical observation of known conjugated structures and applied to new systems.This quantification is referred to as the Woodward-Fieser Rules for eg. - Woodward-Fieser RulesWoodward and the Fiesers performed extensive studies of terpene and steroidal alkenes and noted similar substituents and structural features would predictably lead to an empirical prediction of the wavelength for the lowest energy π → π* electronic transition.This work was distilled by Scott in 1964 into an extensive treatise on the Woodward-Fieser rules in combination with comprehensive tables and examples – (A.I. Scott, Interpretation of the Ultraviolet Spectra of Natural Products, Pergamon, NY, 1964).A more modern interpretation was compiled by Rao in 1975 – (C.N.R. Rao, Ultraviolet and Visible Spectroscopy, 3rd Ed., Butterworths, London, 1975).
- Woodward-Fieser Rules - DienesThe rules begin with a base value for lmax of the chromophore being observed:The incremental contribution of substituents is added to this base value from the group tables:
 acyclic butadiene = 217 nmFor example:
acyclic butadiene = 217 nmFor example:


- Woodward-Fieser Rules – Cyclic DienesThere are two major types of cyclic dienes, with two different base valuesHeteroannular (transoid):
 e = 5,000 – 15,000base λmax = 214
e = 5,000 – 15,000base λmax = 214







No comments:
Post a Comment